The role of molecular chaperones in mediating and controlling intracellular, as well as in vitro protein folding, has broad implications for biotechnology. There is now considerable insight into the possible mechanisms whereby chaperone proteins recognize, stabilize, and release unfolded polypeptide chains in a manner whereby they are able to productively refold. However, there are a number of important, fundamental gaps in the understanding of chaperone action, which remain to be resolved. Among the growing list of chaperone families are the chaperonins G r o E L ( E L ) and G r o E S ( E S ), which have been intensively studied. Knowledge of how misfolded protein substrates physically interact with E L should provide vital clues necessary to unravel the process by which E L mediates their proper folding. The ultimate goal is to determine the mechanism by which E L transforms its substrate proteins and then releases them in a form able to refold to their native conformation.
One of the key issues in establishing a molecular mechanism for E L is to describe in structural terms the conformations of polypeptide substrates when bound to various chaperonin complexes. For example, does a nonnative polypeptide substrate unfold further upon binding to E L? On the other hand, when a chain is released from E L in the presence of the co-chaperonin E S, does it adopt a more folded, or less folded conformation? These questions are difficult to resolve with naturally occurring proteins since they refold so readily when released from chaperonin complexes. One approach to address these issues, however, is to utilize a family of mutationally altered protein substrates that are unable to adopt their native conformation. The nonnative subtilisin variant, PJ9 ( p ), which is unable to refold when released, is one such system that is readily available [ 1 ].
Single-ring E L mutants have been shown to assist in the refolding of nonnative polypeptide chains [ 2, 3 ] and they form unusually stable complexes with E S upon the addition of nucleotides. Capitalizing on these properties, a single-ring E L variant ( s E L) was used to trap p within E L by E S upon the addition of the nucleotides adenosine di- or tri-phosphate ( ADP or ATP ). Small angle neutron scattering ( SANS ) experiments were then performed to examine the structure of s E L / E S / p complexes and investigate changes in p conformation in the presence of both nucleotides.
p was 86 % deuterated ( dp ) so that it contrasted sufficiently with the chaperonin, allowing the contrast variation technique to be used to separate the scattering from the two components bound in the complex. The s E L mutant assured that dp and E S were each bound in a 1:1 stoichiometry with the s E L, providing an advantage over previous SANS experiments [ 4 ] which included mixed stoichiometries of E L and E S.
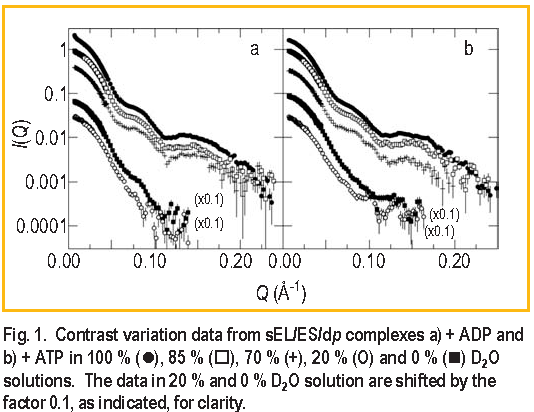
Unlike ATP, ADP does not cause dissociation of dp from E L. If dp is no longer covalently bonded to E L, would its location in the E L / E S complex change? To answer this question, two contrast variation series of measurements, one with ADP and the other with ATP present, were performed on the s E L / E S / dp complex [ 5 ]. Measurements were made in five D2O buffers in each case, as shown in Figure 1. The extensive I ( Q ) data sets in Figure 1 allow the separation of the scattering intensities from each of the components in the complex and also the crossterm intensity. Modeling is then used to try to reproduce these measured intensities.
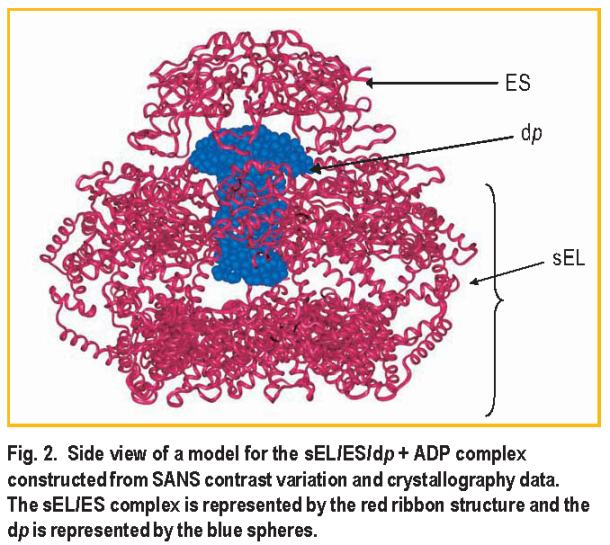
Using this method, the location and approximate shape of dp in the s E L / E S / dp + ADP complex was determined as modeled in Figure 2. The significant result is that the dp component has an asymmetric shape: part of the polypeptide must extend beyond the cavity inside the s E L ring up into the space surrounded by E S. The best fit to the data from both complexes ( i.e., with ADP or ATP ) was achieved using one ring of the E L / E S solution structure of Reference 6 for the s E L / E S component of the complex, one ring of the x-ray structure for E L of Reference 7 for the free s E L, and the x-ray structure for free E S of Reference 8 in a ratio of 75:21:4 by mass.
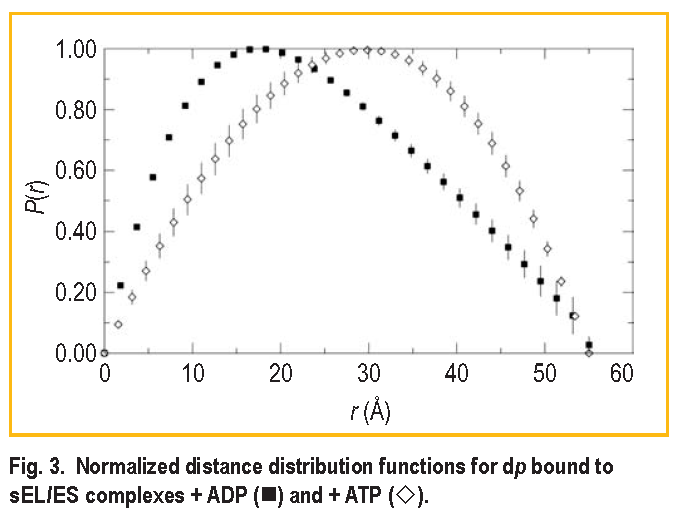
The significant difference in the s E L / E S / dp complex formed from ATP is that the shape of the bound dp molecule changes from an asymmetric shape such as that shown in Figure 2 to a more symmetric shape. Figure 3 shows the distance distribution functions obtained by Fourier inversion of the I ( Q ) for bound dp in both the s E L / E S / dp + ADP and s E L / E S / dp + ATP complexes. The most probable distance increases from approximately 22
to 30
The shape of dp in the presence of ATP is clearly more symmetric, as indicated by the greater symmetry of the distance distribution function. This suggests that dp is transformed into a more expanded form in the ATP complex. However, the symmetry of this curve cannot be used to unambiguously assign a shape to dp in the ATP complex. Because the s E L / E S / dp complex dissociates in solution, which contains about 21 % of s E L / dp complex, it is possible that the distance distribution curve represents a composite of data from dp bound to s E L alone and to the s E L/ E S complex formed from ATP. For the same reason, it is unclear from the data whether dp is still located partially in the s E L cavity. However, what is clear is that there has been a transformation in the shape of dp associated with the complex formed from ATP. The dp shape change was significant enough to be detected as a change in the shape of the scattering from this component, in spite of the fact that dissociation is likely to be present in the sample. A conformational change of dp either was not supported by the complex formed from ADP or was insufficient to generate a lasting change in shape in that case, and dp instead relaxed back to a form close to its original conformation. This important observation reflects the relative ability of ATP to promote refolding of protein substrates relative to ADP.
References:
[1] Z. Lin and E. Eisenstein, Proc. Natl. Acad. Sci. USA 93, 1977 (1996).
[2] I. S. Weissman, Chem. Biol. 2, 255 (1995).
[3] S. G. Burston, J. S. Weissman, G. W. Farr, W. A. Fenton and A. L. Horwich, Nature 383, 96 (1996).
[4] P. Thiyagarajan, S. J. Henderson, and A. Joachimiak, A, Structure 4, 79 (1996).
[5] S. Krueger, S. K. Gregurick, J. Zondlo and E. Eisenstein, J. Struct. Biol. 141, 240 (2003).
[6] R. Stegmann, E. Manakova, M. Rossle, H. Heumann, S. E. Nieba- Axmann, A. Pluckthun, T. Hermann, R. P. May and A. Wiedenmann, J. Struct. Biol. 121, 30 (1998).
[7] K. Braig, Z. Otwinowski, R. Hegde, D. C. Boisvert, A. Joachimiak, A. L. Horwich and P. B. Sigler, Nature 371, 578 (1994).
[8] J. F. Hunt, A. J. Weaver, S. J. Landry, L. Gierasch, and J. Deisenhofer, Nature 379, 37 (1996).
S. Krueger
NIST Center for Neutron Research
National Institute of Standards and Technology
Gaithersburg, MD 20899-8562S. K. Gregurick
University of Maryland, Baltimore County
Baltimore, MD 21250J. Zondlo and E. Eisenstein
Center for Advanced Research in Biotechnology
University of Maryland Biotechnology Institute
Rockville, MD 20850
Back to FY2003 HTML main page
Go to previous article
Go to next article
To view all symbols correctly, please download Internet Explorer 6 or Netscape 7.1