We have been engaged in an interdisciplinary materials research effort to develop new classes of biomaterials that are constructed via designed molecular selfassembly [ 1 ]. These materials use specific polypeptides that are designed a priori to self-assemble into targeted nano- and microscopic structures. Using amino acids as the fundamental material building blocks, one can potentially engineer materials having targeted biological functions such as in vivo therapeutic delivery or tissue scaffolding. Materials can be designed whose morphology is responsive to specific environmental cues. The ability to actively manipulate material morphology can lead to “smart” materials whose structure and consequent biological function is responsive to environmental cues. Predictable control of material morphology would be a significant advance in the development of functional biomaterials because it would provide active control of biological properties that are dependent on an aggregate’s shape or size. An example of molecular design for a specific application would be in the delivery of drugs and genes, where the shape of the delivery assembly favors selective interactions with different biological surfaces, e.g., an active gating mechanism induced by a predicted external stimulus.
Polypeptides, composed of amino acids each of which imparts a unique structural and/or functional characteristic to the polymer molecule, provide an immense molecular toolbox from which one can construct functional materials via self-assembly. The primary tools exploited are the conformational propensities of individual amino acid residues to adopt desired secondary structures or shapes and the tendency of the appropriately designed secondary elements to self-associate ( Figure 1 ). It is now possible to design peptides that will fold into stable secondary structures such as
-helices ( rod-like polymers that typically form nematic liquid crystals in solution and pack as hexagonal arrays of rods in the solid-state ) and
-sheets ( crystalline arrays of extended chains, mainly stabilized by H-bonding ). It is also possible to design polypeptides that, in addition to intramolecular folding, undergo self-assembly to construct higher order structures.
Currently we are studying the self-assembly behavior of both charged and neutral amphiphilic di- and tri-block copolypeptides. Unlike membrane-forming lipids and surfactants, the neutral copolypeptides form robust bilayer membranes in aqueous solutions over a wide range of molecular weights and molar ratios. This has been observed in amphiphilic, nonionic, completely
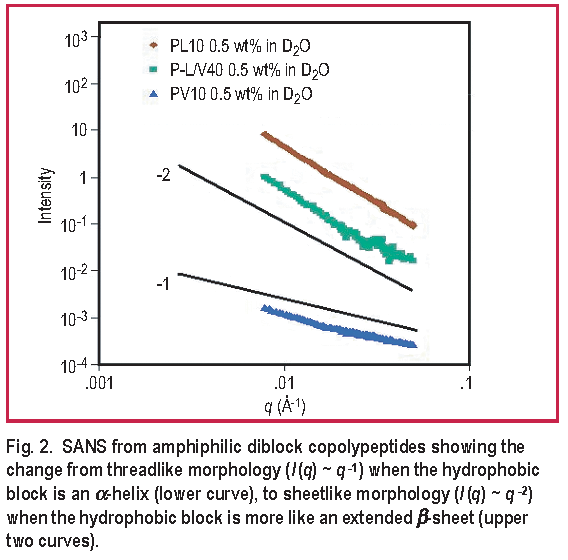
A SANS study of secondary structure effects shows a large transition in the assembled structure induced by changing the minority hydrophobic block from an
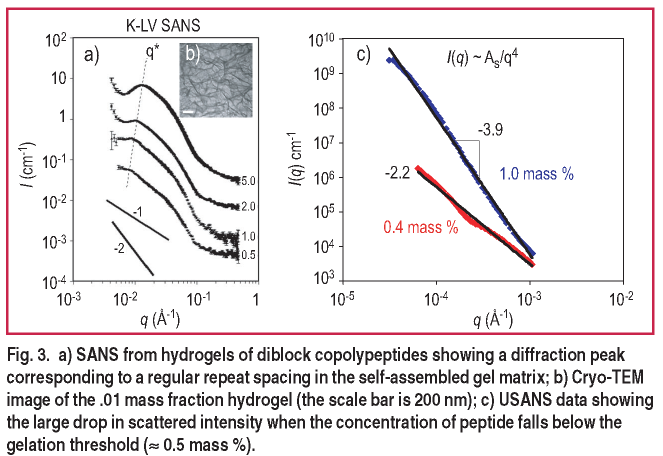
In addition to the bilayer/membrane forming nonionic polypeptides, hydrogel formation of charged, amphiphilic block polypeptides is observed. By removing the PEG side chain from poly-L-lysine, one obtains a polycationic lysine block at neutral pH. Block polypeptides of lysine and leucine form strong hydrogels at low molecular weights ( 20k g/mole ) and concentrations ( < 0.006 mass fraction ), both an order of magnitude lower than observed in traditional polymeric hydrogel formation. The structural characterization of these unique gels is crucial to uncovering their gelation mechanism and in the design of new gel formers with the added control/environmental responsiveness discussed above. Gelation occurs with a variety of hydrophobic block secondary structures but with varying degrees of strength [ 2 ]. Both SANS and USANS measurements at the NCNR have been indispensible for probing the global nano- and microstructure of these porous hydrogel materials.
In some cases, the self-assembled gels exhibit a characteristic spacing in the underlying scaffold. An example of this is observed in Figure 3 where the correlation peak shifts to lower q with decreasing concentration, indicating that the regular structure persists even as the characteristic spacing increases [ 3 ]. On the microscale, the hydrogels exhibit clear smooth surface scattering with I ( q ) ~ q-4 as determined by USANS ( Figure 3c ). When the peptide concentration drops below the gelation threshold (
0.005 mass fraction ) the material becomes a viscous liquid, and the USANS scattering drops significantly in slope indicating the loss of well-defined microstructure in the materials [ 3 ].
We have also investigated the dynamics of the gelation process [ 1 ]. In a rheometer, the gel structure was disrupted by large amplitude oscillations, but recovered 90 % of full strength within seconds of cessation of shear. The low mass fraction of material in the polypeptide hydrogels, combined with their recovery properties and microporous structure, allows them to fill an advantageous and unique niche between conventional polymer and surfactant hydrogels. Their peptide backbone also imparts to these materials some of the advantageous features of proteins ( such as degradability and functionality ), which makes them attractive for biomedical applications.
References:
[1] A. P. Nowak, V. Breedveld, L. Pakstis, D. J. Pine, D. J. Pochan, T. J. Deming, Nature 417, 424 (2002).
[2] J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. M. Pakstis, J. Gill, J. Am. Chem. Soc. 124, 15030 (2002).
[3] D. J. Pochan, L. Pakstis, B. Ozbas, A. K. Nowak, T. J. Deming, Macromolecules, 35, 5358 (2002).
[4] J. Kopecek, Nature 417, 388 (2002).
D. J. Pochan, J. P. Schneider, L. Pakstis, B. Ozbas
University of Delaware, Newark, DEA. P. Nowak and T. J. Deming
University of California at Santa Barbara
Santa Barbara, CA
Back to FY2003 HTML main page
Go to previous article
Go to next article
To view all symbols correctly, please download Internet Explorer 6 or Netscape 7.1
![Figure 1. a) Schematic of peptide monomers chemically bonded to form diblock, triblock and random copolymers; b) Amphiphilic copolypeptides self-assembled in micelles; c) Copolypeptides physically linked to form a hydrogel; and d) typical copolypeptide chain architectures [4].](figure1.png)