Proteins are long macromolecular chains consisting of amino acid repeat units that, in the native state, adopt a well defined and intricately folded three-dimensional conformation. The ability to predict a priori the native three-dimensional structure of a given amino acid sequence, the so called “protein folding problem,” is essential for the full promise of genetic therapies to come to fruition (Refer to reference 1). In fact, many apparently unrelated diseases (e.g., Alzheimer’s, Mad Cow, cystic fibrosis, and even many cancers) result from protein folding gone wrong.
In order to obtain detailed knowledge of the protein folding mechanism, extensive characterization of the relevant interactions, structure and dynamics is required at each step of the protein-folding pathway. To this end, researchers have been investigating the properties of proteins in the native, partially unfolded and the completely unfolded states to obtain “snapshots” of the relevant behavior of intermediates along the folding pathway, yielding information intended to facilitate rationalization of folding mechanisms.
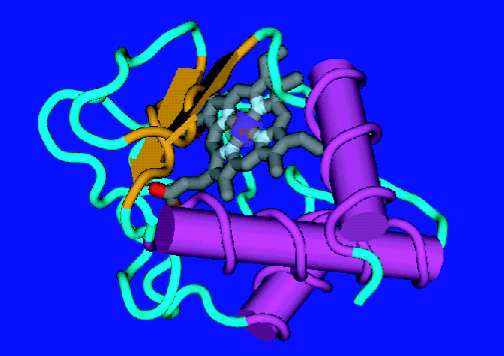
| Figure 1. The native state of cytochrome c. |
To deepen our understanding of the molecular motions involved in protein folding and, hence, the role of dynamic behavior on the overall folding mechanism, we employed quasielastic neutron scattering (Q E N S) as a dynamical probe of several folded states of the electron transfer protein cytochrome c (see Figure 1). Q E N S provides a method for directly probing the internal dynamics of biomolecules on the pico second time scale and Angstrom length scale. In this technique neutrons exchange energy and momentum with the nuclei of the sample, allowing the forms, geometry, amplitudes and time-dependence of the motions involved to be determined. By making use of the large incoherent cross section of hydrogen nuclei and the capabilities of the Disk-Chopper Spectrometer at approximately 32 micro electron volts resolution it was possible to collect data on five different cytochrome c folded states (in order of increasing unfolding): native (N), salt induced molten globule (M G), alkaline denatured (D, sub A L K), acid denatured (D, sub acid) and guanidine hydrochloride unfolded (U).
In a typical Q E N S experiment of this sort, a monochromatic beam of neutrons is scattered by a buffered solution containing the protein, the contribution of the scattering from the buffer is removed, and the resulting profile is considered to be that resulting from just the protein (Refer to references 2 and 3). Figure 2 depicts the general neutron scattering profile from a protein in solution, illustrating the distinct scattering from global (bulk translational and rotational diffusion) and internal (protein side-chain) dynamics. A profile of this sort is analyzed with a two Lorentzian fit model — a narrow Lorentzian to fit the global motion and a broad Lorentzian to fit the internal motions. The two Lorentzian fit model permits the average ensemble time scale of global and internal side-chain motions to be ascertained separately.
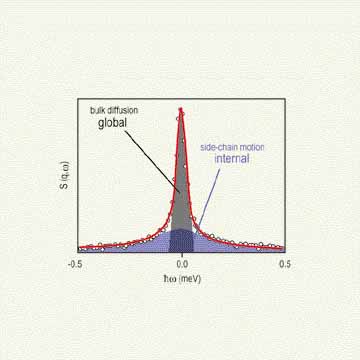
| Figure 2. Illustration of the general quasielastic neutron scattering profile of a protein in solution indicating the narrow scattering from the global diffusion and the broad scattering due to the internal side chains of the amino acid residues. |
The half width at half maximum of both Lorentzian fit functions, corresponding to the global and internal motions as a function of scattering vector squared (q squared) are given in Figure 3. An effective diffusion coefficient (D sub e f f) is determined from the slope of capital gamma sub global as a function of q squared. As evident from the tabulated results, D sub e f f generally decreases as the protein unfolds reflecting the correspondingly higher specific molecular volume of non-native folding states. The exception lies with the acid denatured state that, due to the strong electrostatic interactions present in the system, shows a near native D sub e f f..
A decrease in the relative time scale of the internal motions is observed as a function of protein unfolding. It is believed that the rather close-packed native structure provides more sharply defined, short-range interactions between the proximate side chains of the amino acid residues. Upon unfolding of the protein, these interactions are diminished by the increased separation between side chains and small, weakly interacting buffer molecules between them.
The results of this study demonstrate the type of dynamic information that can be determined from Q E N S experiments performed on a protein in several different structural states. As the nature of the unfolding medium displays a critical role in the internal dynamics of the unfolded states of cytochrome c, any attempts to correlate features between different folding states should take this into account. In combination with other characterization techniques, information of this sort will prove beneficial to the understanding and development of accurate protein folding mechanisms.
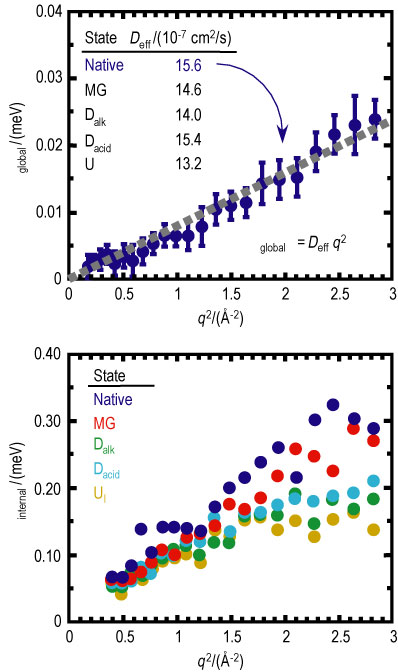
| Figure 3. Plot of the half width at half maximum of the global (top, native only) and internal (bottom) Lorentzian as a function of scattering vector squared. The tabulated effective diffusion coefficients are given for each folded state of cyt c. |
References
[1] A. R. Dinner, M. Karplus, Angew. Chem. Int. Ed. 40, 4615 (2001).
[2] Z. Bu, D. A. Neumann, S-H. Lee, C. M. Brown, D. M. Engelman, C. C. Han, J. Mol. Biol. 301, 525 (2000).
[3] J. Perez, J.-M. Zanotti, D. Durand, Biophys. J. 77, 454 (1999).
Author names
Author affiliation
Insitution
Town, ST zzzipp
Authors
A. Pivovar and D. A. Neumann
NIST Center for Neutron Research
National Institute of Standards and Technology
Gaithersburg, MD 29899-8562
Back to FY2002 HTML main page
Go to next article
To view all symbols correctly, please download Internet Explorer 6 or Netscape 7.1